
Des pompons pour mieux comprendre le séquençage de l'ADN publié le 09/02/2022
Modélisation de la méthode de Sanger
Le principe du séquençage mis au point par Frederick Sanger en 1977 est illustré de diverses façons dans tous les nouveaux manuels d’enseignement scientifique de première. Après avoir étudié toutes ces illustrations et passé plusieurs longues minutes à expliquer la méthode en cours magistral, avec le support qui me semblait le plus pertinent, il s’est avéré qu’un nombre non négligeable d’élèves n’avaient finalement pas compris ce qui se passait et étaient incapables de lire une portion d’un gel de séquençage.
J’en ai donc déduit qu’une modélisation pouvait être utile. À priori, quand j’utilise l’activité proposée dans cet article, beaucoup plus d’élèves réussissent l’évaluation de fin de chapitre.
Place de l’activité dans la progression
Nous sommes dans le Thème 1 : La Terre, la vie et l’organisation du vivant, dans la partie : Transmission, variation et expression du patrimoine génétique, et dans le chapitre : l’histoire humaine lue dans son génome.
La méthode de Sanger concerne la biologie moléculaire de l’ADN, je l’aborde donc après avoir travaillé en classe les chapitres majeurs de génétique, c’est à dire :
- La division cellulaire (réplication, PCR, mitose, méiose)
- Les mutations et la variabilité génétique.
- L’expression du patrimoine génétique.
Mise en œuvre de l’activité
- La méthode de séquençage n’est pas étudiée pour elle même, elle est intégrée dans un TP numérique où les élèves doivent réfléchir sur ce que nous ont légué (au niveau génétique) les Dénisoviens.
Dans le TP proposé, seul le premier objectif pédagogique traite du séquençage. Le deuxième objectif repose sur un fichier d’arbre moléculaire disponible sous Phylogène (ADN mitochondrial de quelques homininés). Le troisième objectif utilise des fichiers ressources disponibles sur le site Acces de Lyon : génétique de l’adaptation à l’altitude des Tibétains. Les élèves exploitent la comparaison de séquences du gène EPAS1 avec Anagène.
- Le point de départ de l’activité s’appuie sur un document du Manuel Magnard (page 66). L’ADN d’une phalange fossile de Dénisovien, datée de -41000 ans, est extrait puis amplifié par PCR. La technique de PCR a été étudiée dans les cours précédents. Chaque binôme va donc se retrouver avec un fragment théorique différent de 10 nucléotides, issu de l’amplification. Le séquençage de chaque binôme est ensuite reporté dans un tableau récapitulant les résultats de la classe.
- Quelques informations sur la méthode de Sanger, données en début de séance
L’ADN amplifié par PCR est placé dans un milieu contenant les 4 nucléotides A T G C et de l’ADN polymérase. Des amorces aléatoires permettent de fabriquer de nombreux fragments d’ADN complémentaires.
Environ 0,2 % des nucléotides du milieu sont marqués par fluorescence, chaque nucléotide a « sa » couleur. Quand l’ADN polymérase utilise par hasard un nucléotide fluorescent, les fragments d’ADN incorporant ce nucléotide seront également fluorescents.
Les précisions fournies ci-dessus sont volontairement succinctes. C’est en fin de modélisation que les élèves auront à réfléchir sur la signification du protocole manipulatoire que je leur impose.
Un gel de séquençage est également vidéo projeté
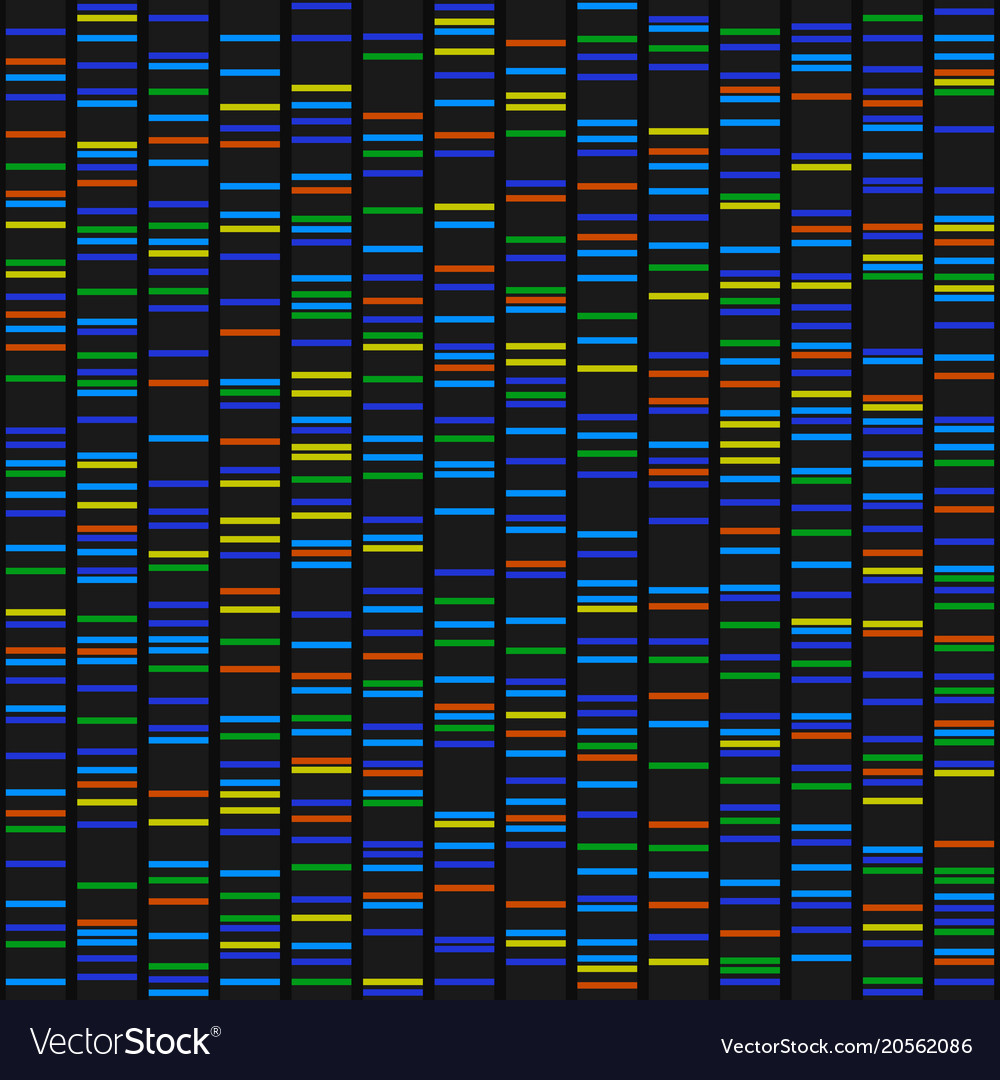
Modélisation de la méthode de Sanger

- Prendre 20 pompons blanc + 4 pompons de couleur : 1 vert + 1 rouge + 1 jaune + 1 bleu.
- Les placer dans un grand verre cartonné puis réaliser une série de tirages au sort, un pompon à la fois.
- À chaque fois que vous tirez un pompon de couleur, le tirage s’arrête et vous notez le nombre de pompons blancs (nucléotides non fluorescents) ainsi que la couleur du dernier pompon (nucléotide fluorescent).
- Reportez votre résultats dans le tableur disponible.
- Tous les pompons sont remis dans le verre après chaque tirage.
- Quand un deuxième tirage s’arrête sur un nombre de pompons déjà tiré au sort, vous ignorez le résultat.1
- Tirer au sort autant de fois que nécessaire pour obtenir des pompons de couleurs sur les positions n°1 à n°10 nucléotides.
- Vous pouvez par contre obtenir un nombre de pompons supérieurs à 10 nucléotides.
- Appeler l’enseignant quand vous avez réalisé 20 tirages successifs (pour vérification des résultats et obtention des consignes pour terminer le séquençage.2).
- Après validation, saisir votre séquence d’ADN dans le tableau commun à la classe.
- Construire la séquence d’ADN la plus longue possible à l’aide des résultats de tous les binômes.
Les deux tableurs pour reporter les résultats :
Exploitation des résultats
Voici un exemple de tableau obtenu par un binôme :
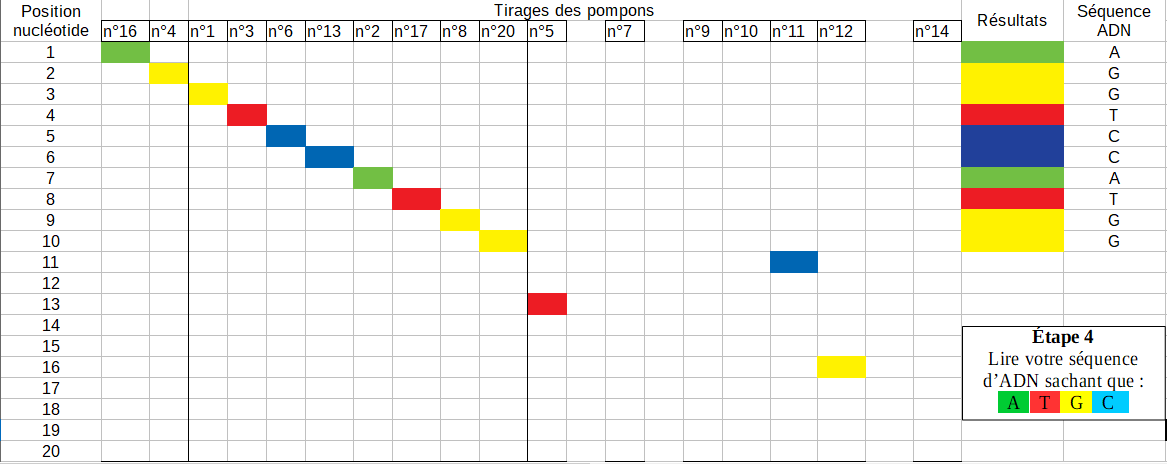
Les élèves sont bien allés jusqu’au vingtième tirage, leur tri de colonnes semble correct (partie gauche du tableau). Ils ont supprimé quelques tirages redondants mais les colonnes vides dans la partie droite du tableau révèlent peut-être une méthode numérique originale pour les copier-coller des colonnes ou le report des résultats.
Le code couleur A = vert, T = rouge, G = jaune, C = bleu, donné après vérification du suivi de consignes sur le protocole manipulatoire, leur a permis de trouver la séquence suivante : AGGTCCATGG.
Le hasard du tirage au sort fait qu’ils ont aussi trouvé des nucléotides au delà de la dixième position (tirages n°5, n°11, n°12).
Voici les résultats des 2 groupes de la classe, soit 2 fois 9 séquences d’ADN :
Les élèves se déplacent et renseignent le tableau récapitulatif à tour de rôle sur un ordinateur dédié à la récupération des résultats du groupe. Ce tableau est sauvegardé par l’enseignant sur l’espace de travail collaboratif de l’ENT et les élèves le récupèrent pour assembler, à la manière des dominos, une séquence d’ADN la plus longue possible.
Cette opération permet de comprendre comment on travaille sur le puzzle des multiples séquençages pour obtenir un génome complet.
Cette année, avec les fragments du groupe n°1, il a été possible de séquencer un fragment de 34 nucléotides.
Avec le groupe n°2, nous obtenons deux fragments séparés, un de 17 et un de 18 nucléotides.

Rien n’empêche l’enseignant de regrouper toutes les séquences dans un même tableau pour poursuivre le travail d’assemblage en exercice, corrigé lors de la prochaine séance.
Bien évidemment, il faut qu’au moins deux fragments s’assemblent dans le groupe pour exploiter cette dernière partie du raisonnement.
Expliquer la méthode de séquençage
C’est une activité réalisée individuellement, les binômes rédigent chacun leur texte explicatif sans communiquer entre-eux. Un document papier illustratif est distribué en guise d’aide au raisonnement (document modifié à partir du site Futura-Sciences.com).

On s’attend a obtenir plusieurs phrases, une par étape sur le schéma, et surtout l’élément clé de la compréhension de la méthode, à savoir que l’incorporation d’un nucléotide fluorescent bloque la synthèse de l’ADN. La technique d’électrophorèse a été abordée dans les TP précédents (chapitres de la réplication de l’ADN avec la PCR et de l’expression du patrimoine génétique) mais une précaution doit être prise en corrigeant l’activité. L’électrophorèse trie les fragments du plus long (au sommet du gel) au plus court (fin du gel) et il n’est pas rare que les élèves lisent à l’envers la séquence génétique quand leur attention n’a pas été attiré sur ce point précis. L’idéal serait de faire correspondre le sens de migration du gel de séquençage dans l’évaluation de fin de chapitre, avec le sens de migration imposé dans le TP. Il faut dans tous les cas rappeler que la lecture d’un gel de séquençage se fait dans le sens inverse de la migration électrophorétique.
Ainsi, le document illustratif donné en fin de séance peut semer la confusion, car dans le tableau de résultats des binômes, la migration se ferait de bas en haut, tandis que sur le schéma, elle s’effectue de haut en bas.
(1) C’est un point délicat de la modélisation, normalement, il ne devrait pas être possible que plusieurs nucléotides tombent sur la même position. Il faut expliquer que cette astuce est une limite du modèle et ne correspond pas à la réalité de la méthode de séquençage.
(2) C’est à ce moment que je donne la correspondance entre les couleurs de pompons et les nucléotides